 Электронная подпись (ЭЦП) в Твери: основы и применение
Электронная подпись (ЭЦП) в Твери: основы и применение  Как найти идеального сотрудника?
Как найти идеального сотрудника?  Самозанятый в Тинькофф
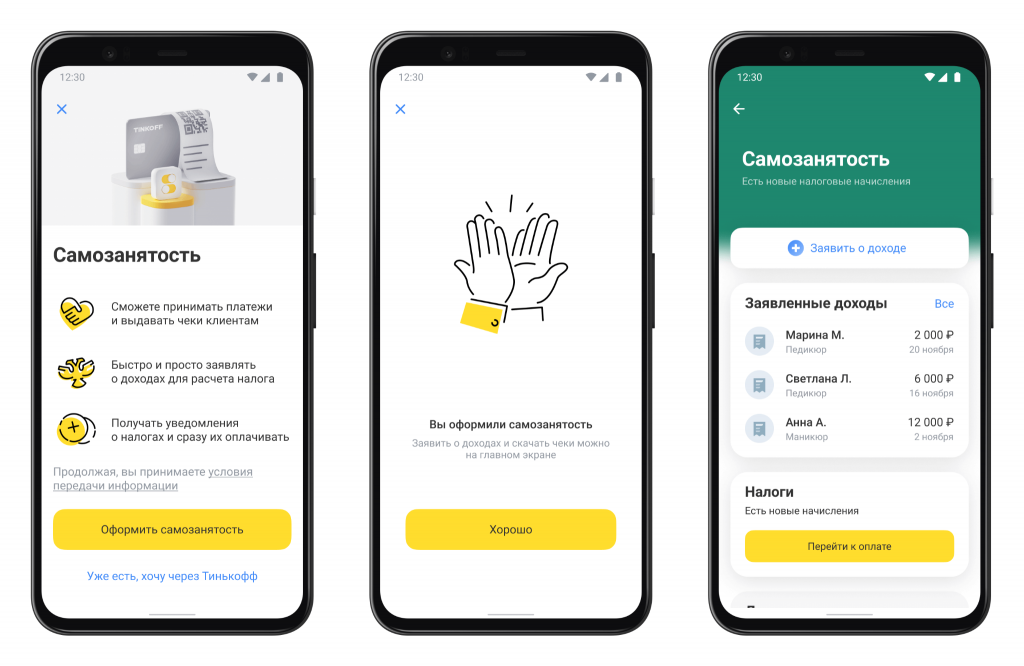
Самозанятый в Тинькофф  Самозанятые в Тинькофф: возможности и условия для фрилансеров и индивидуальных предпринимателей
Самозанятые в Тинькофф: возможности и условия для фрилансеров и индивидуальных предпринимателей  Тимур Турлов: казахстанский феномен в мире финансов
Тимур Турлов: казахстанский феномен в мире финансов 



/imgs/2017/05/12/13/755177/14b8eead29f9fdfd818446f9cf731f5bef5c7d62.jpg)


Электронная подпись (ЭЦП) в Твери: основы и применение
Электронная подпись (ЭЦП) становится неотъемлемой частью современного документооборота, упрощая процессы подписания документов, сокращая время их обработки и увеличивая уровень безопасности....
Электронная подпись (ЭЦП) становится неотъемлемой частью современного документооборота, упрощая процессы подписания документов, сокращая время их обработки и увеличивая уровень безопасности....

Рассказывает Дина ГУбайдулина - основатель компании по созданию вовлеченных команд «Skills Crew». Каждый предприниматель рано или поздно сталкивается с наймом...

Что такое самозанятость? Самозанятость - это форма занятости, при которой человек работает на себя и получает доход от своей деятельности....
Самозанятость в России – это относительно новая форма ведения бизнеса, которая предоставляет фрилансерам, мастерам индивидуальных услуг и небольшим предпринимателям возможность...

Бизнес с человеческим лицом: как Тимур Турлов меняет мир к лучшему Тимур Турлов с юности интересовался финансами. В 21 год,...

Зачем осваивать управленческий учет? Овладение знаниями управленческого учета является неотъемлемой частью работы менеджера или руководителя. Понимание принципов и методов управленческого...

Центр сертификации продукции и услуг в Москве https://artalix.ru/ - это организация, занимающаяся проверкой и удостоверением соответствия товаров и услуг установленным...

Бурение скважин на воду в Чувашии – это актуальная услуга как для частных домовладельцев, так и для предприятий, стремящихся обеспечить...

Прозрачная пленка – это универсальный материал, который находит своё применение в самых разных сферах, начиная от бытовых нужд и заканчивая...
Отзыв о работе с Брокером Seneca Corporate Для начала хочу кратко рассказать, что это за платформа. Seneca Corporate — биржа, предоставляющая услуги...